Research Projects



Project 1
Investigates the cellular and molecular mechanisms that underly dystroglycanopathies using state-of-the-art biochemical, cell biological and glycobiological approaches, recombinant enzymes, and studies in a dystroglycanopathy mouse model. Mechanistic insights into the dystroglycanopathies will provide a rationale for the design of novel diagnostic and therapeutic strategies, as well as approaches to monitoring therapeutic engagement and efficacy.
Project 2
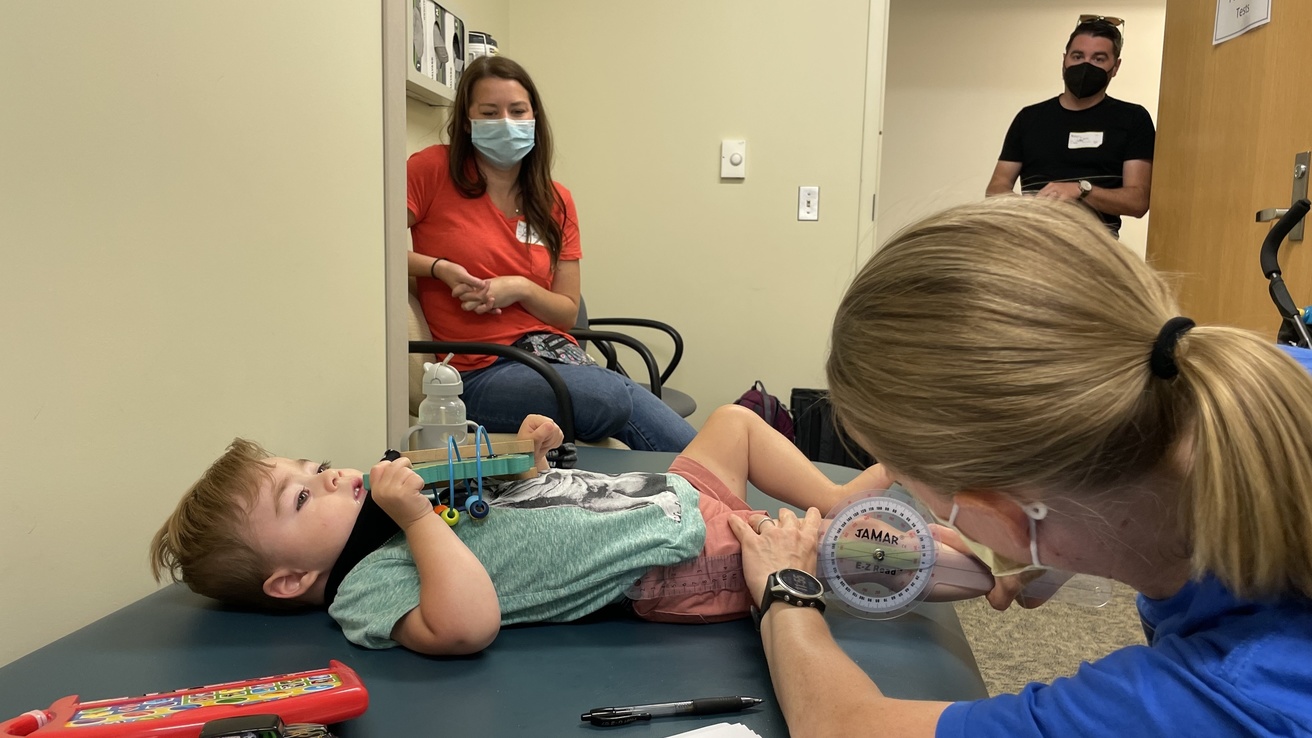
Characterizes the natural history of the dystroglycanopathies using established and evolving clinical measures to optimize clinical care as well as inform and enhance clinical trial design. Project 2 will refine the natural history of FKRP-related dystroglycanopathy derived from an established, unique cohort of patients and extend follow up of non-FKRP, dystroglycanopathy genotypes to identify cohorts that share similar rates of motor progression who might be studied together in gene non-specific clinical trials. Candidate proteomic biomarkers in blood or urine for the full spectrum of dystroglycanopathy genotypes will be validated and related to disease status.